

Graduate Fields: Plant Pathology & Plant-Microbe Biology; Plant Biology
The development of high throughput technologies has given rise to a wealth of information at system level including genome, transcriptome, proteome and metabolome. However, it remains a major challenge to digest the massive amounts of information and use it in an intelligent and comprehensive manner. To address this question, Dr. Fei’s group has focused on developing computational tools and resources to analyze and integrate large scale “omics” datasets,” which help researchers to understand how genes work together to comprise functioning cells and organisms.
See more...
Development of online databases to facilitate data distribution, analysis, mining and integration
- Tomato Functional Genomics Database
- Tomato Epigenome Database
- Cucurbit Genomics Database
- RadishBase
- Kiwifruit Genome Database
- SpinachBase
- Whitefly Genomics Database
- Chinese Tomato Virome
- Pan-African Sweet Potato Virome
Development of computational tools for omics data analysis
- Plant MetGenMAP – a web-based tool for comprehensive mining and integration of gene expression and metabolite changes in the context of biochemical pathways.
- iAssembler – A de novo assembly package for transcriptome sequences generated using 454 or Sanger platforms
- iTAK – A package to identify and classify plant transcription factors and protein kinases.
- VirusDetect – An automated pipeline for efficient virus discovery using deep sequencing of small RNAs.
Application of NGS technologies and bioinformatics in crop improvement
During the past several years, significant progresses have been made regarding the DNA sequencing technologies. As a result, several next-generation sequencing (NGS) platforms, such Illumina HiSeq, have received wide applications due to their high throughput and low cost. We are interested in using NGS technologies to investigate genomes, epigenomes and transcriptomes of several economically important crops including tomato, cucurbits, sweetpotato, and fruit tree crops, to facilitate the understanding of the evolution and regulatory networks of important agronomical traits. We are also using NGS technologies to perform large-scale virus survey for crops like sweet potato and tomato, in an effort to understand global virus diversity, distribution and evolution in important food crops.
Inferring gene regulatory networks
Living cells are the product of gene expression programs involving regulated transcription of thousands of genes. How a collection of transcriptional regulatory factors associates with genes during specific biological processes or under specific environmental conditions can be described as a gene regulatory network. We are interested in developing new algorithms to infer gene regulatory networks by integrating datasets from various different sources, including gene expression data, metabolomics data, promoter sequences, and microRNA information.


-

-

-

-

Study Reveals Role of Allele Dosage in Improving Sweetpotato Traits
Sweetpotatoes are an agricultural powerhouse that feeds millions globally. However, their complex genetics make it challenging for breeders to understand and improve traits like yield, disease resistance, and nutritional content. […] Read more » -

-

-

-

-
-

-

-

-

Controlling insect pests by targeting genes acquired from other species
Killing crop-damaging insects by targeting genes essential to their survival is a promising approach to pest control. Because essential genes are often conserved across multiple insect species, the challenge is […] Read more » -

Wild tomato genome will benefit domesticated cousins
Wild relatives of crops are becoming increasingly valuable to plant researchers and breeders. During the process of domestication, crops tend to lose many genes, but wild relatives often retain genes […] Read more » -

BTI Welcomes Summer Student Interns
On May 31, Boyce Thompson Institute welcomed 41 of the country’s brightest undergraduate students from universities around the country to experience the life of a researcher for 10 weeks. Ten […] Read more » -

Genetic discoveries could improve spinach’s disease resistance and palatability
Most of us are familiar with “spinach teeth,” the harmless but gritty-chalky mouthfeel caused by the vegetable. A team of researchers from Boyce Thompson Institute (BTI) and six Chinese universities […] Read more » -

Gene discovery may help peaches tolerate temperature stress
A BTI-led team has identified genes enabling peaches and their wild relatives to tolerate stressful conditions – findings that could help the domesticated peach adapt to atmospheric disruptions. The study, […] Read more » -

Tomato’s Wild Ancestor Is a Genomic Reservoir for Plant Breeders
Thousands of years ago, people in the region now known as South America began domesticating Solanum pimpinellifolium, a weedy plant with small, intensely flavored fruit. Over time, the plant evolved […] Read more » -

Silk Road Contains Genomic Resources for Improving Apples
The fabled Silk Road – the 4,000-mile stretch between China and Western Europe where trade flourished from the second century B.C. to the 14th century A.D. – is responsible for […] Read more » -

Congratulations Spring 2020 Graduates!
We are pleased to announce that six BTI researchers received their degrees from Cornell University this spring. Congratulations to our newest alumni: Jason Hoki, Schroeder lab, PhD in Chemistry & […] Read more » -
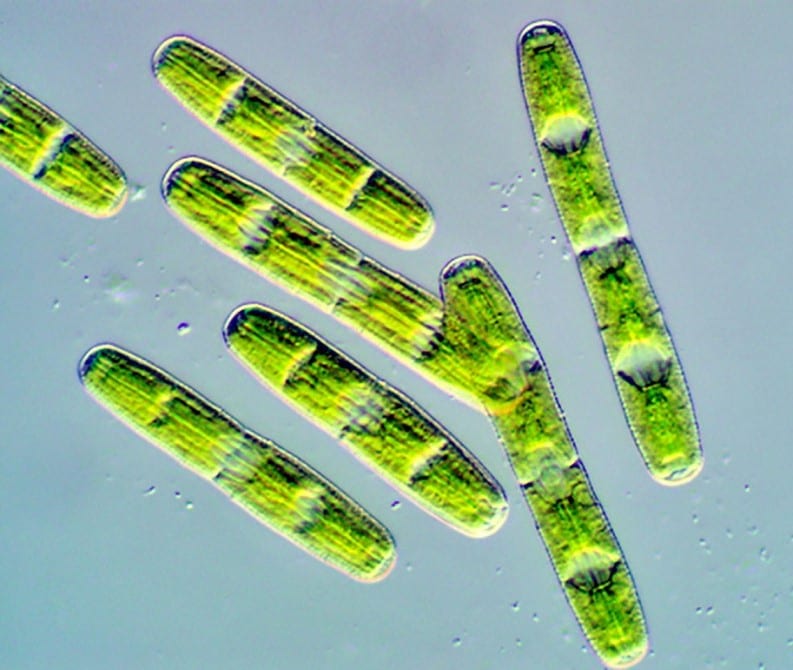
Algal genome provides insights into first land plants
In order to shift from water to land – a transition that still puzzles scientists – plants had to protect themselves from drying out and from ultraviolet (UV) radiation, and they […] Read more » -

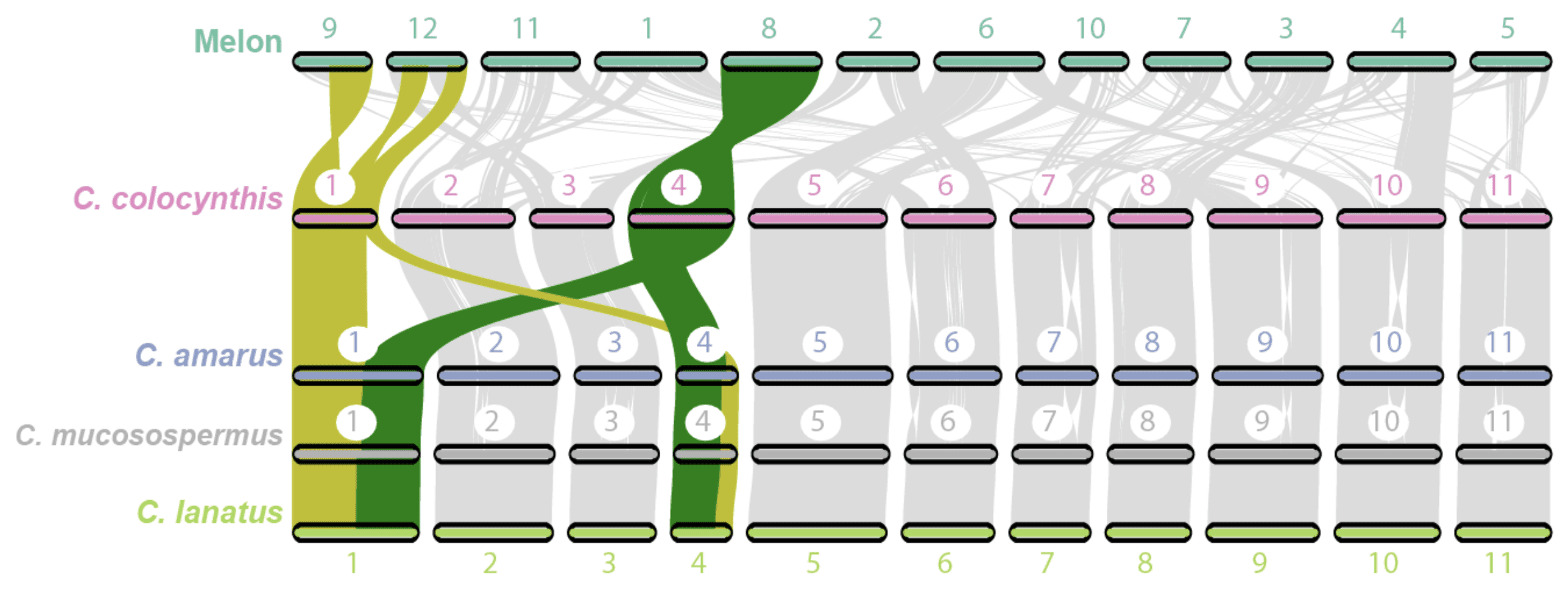
Harvesting Genes to Improve Watermelons
When many people think of watermelon, they likely think of Citrullus lanatus, the cultivated watermelon with sweet, juicy red fruit enjoyed around the world as a dessert. Indeed, watermelon is […] Read more » -
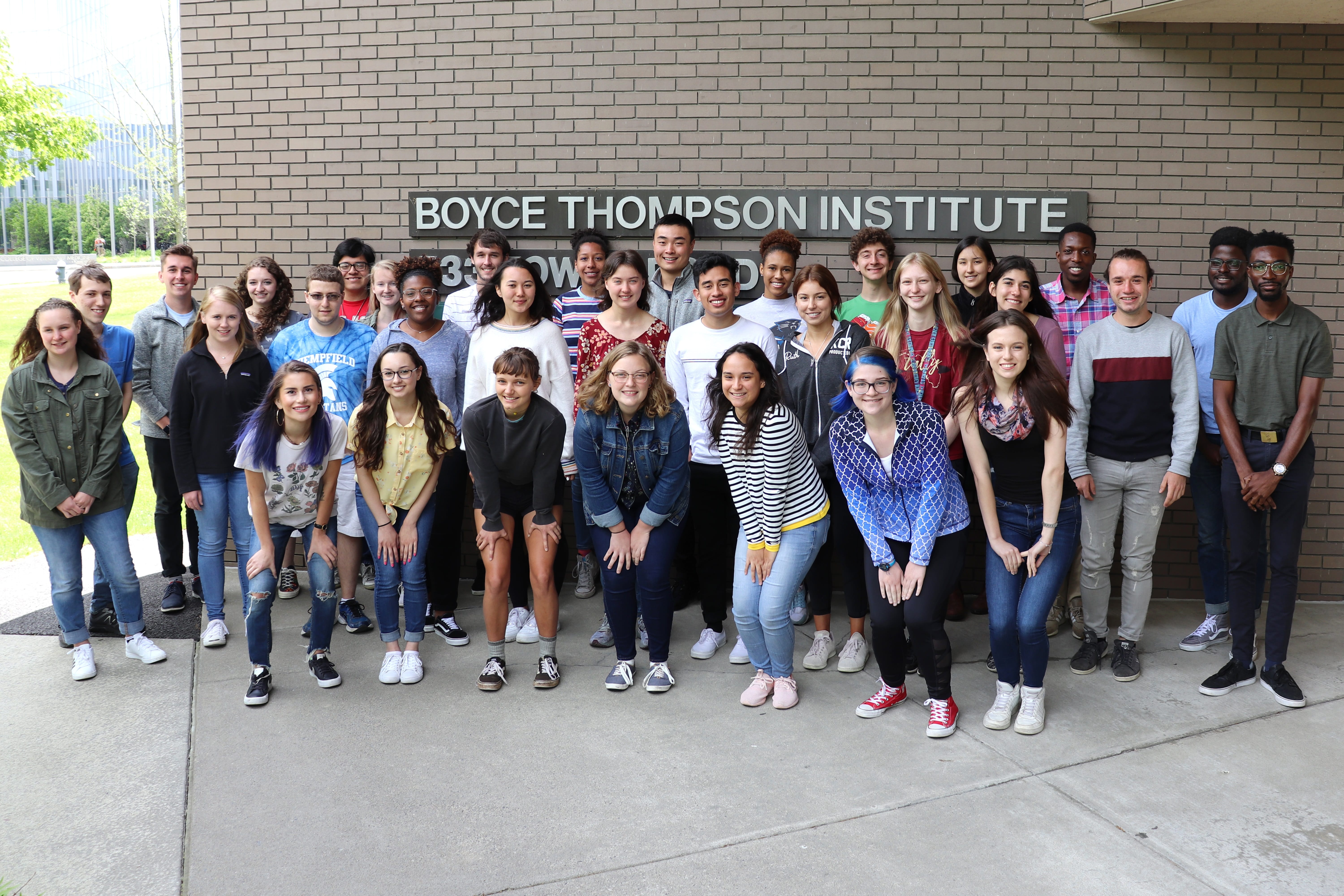
-

BTI Scientists Create New Genomic Resource for Improving Tomatoes
Tomato breeders have traditionally emphasized traits that improve production, like larger fruits and more fruits per plant. As a result, some traits that improved other important qualities, such as flavor […] Read more » -
BTI Promotes Faculty Member Fei
David Stern, president of the Boyce Thompson Institute (BTI), is delighted to announce that faculty member Zhangjun Fei has been promoted to Full Professor on February 27, 2019. Fei was […] Read more » -

New CRISPR database to catalyze collaborations
Recently developed gene editing tools like CRISPR/Cas enable scientists to figure out the functions of myriad plant genes. While these studies could eventually lead to the creation of crops with […] Read more » -

Orange is the new white: New sweetpotato data is something to be thankful for
The genome sequences of I. trifida and I. triloba can be used as robust references to facilitate sweetpotato breeding. The genomic resources developed in this study set the stage for increased rates of genetic gains for key traits such as yield, resistance to disease, and high beta-carotene. Read more » -
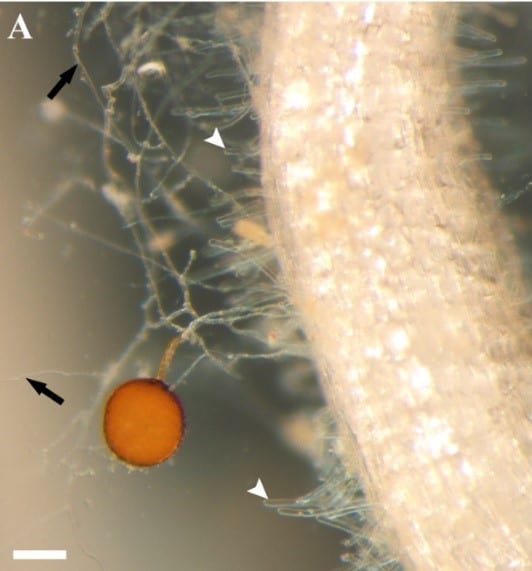
Back to our roots: Insights from genomes of a plant-associated fungus and its bacterial endosymbionts
In an article published this month in the journal New Phytologist, researchers at the Boyce Thompson Institute and the National Center for Genome Resources describe the genome sequences (DNA sequences), […] Read more » -

Pear genomes show evidence of independent domestication in Asia and Europe
Researchers from the Boyce Thompson Institute (BTI) and partnering institutions in China, the U.S., and New Zealand, report their findings on the domestication of the pear in Genome Biology. Read more » -

New ‘Tomato Expression Atlas’ dives deep into the fruit’s flesh
Researchers at BTI, Cornell and USDA published a spatiotemporal map of gene expression across all tissues and developmental stages of the tomato fruit – the genetic information underlying how a fruit changes from inside to out as it ripens. Their data is available in the new Tomato Expression Atlas (TEA). Read more » -

Bottle gourd genome provides insight on evolutionary history and genetic relationships of cucurbit crops
In their findings, researchers compared the sequenced bottle gourd genome to those of other cucurbit species, allowing them to reconstruct the ancient genomic history of the Cucurbitaceae family. Read more » -

Pumpkin genomes sequenced revealing uncommon evolutionary history
For some, pumpkins conjure carved Halloween decorations, but for many people around the world, these gourds provide nutrition. Scientists at Boyce Thompson Institute (BTI) and the National Engineering Research Center for Vegetables in Beijing have sequenced the genomes of two important pumpkin species, Cucurbita maxima and Cucurbita moschata. Read more » -

New genomic insights reveal a surprising two-way journey for apple on the Silk Road
New research out of Boyce Thompson Institute reveals surprising insights into the genetic exchange along the Silk Road that brought us the modern apple. Read more » -

Newly-published spinach genome will make more than Popeye stronger
Today in Nature Communications, researchers from BTI and the Shanghai Normal University report a new draft genome of Spinacia oleracea, better known as spinach. Additionally, the authors have sequenced the transcriptomes (all the RNA) of 120 cultivated and wild spinach plants, which has allowed them to identify which genetic changes have occurred due to domestication. Read more » -
VirusDetect: a new pipeline for virus identification
The Fei lab releases VirusDetect, an automated bioinformatics pipeline that efficiently detects viruses and viroids from large-scale, small RNA datasets. Read more » -

SolGenomics Meeting Has Newest Advances in Nightshades
Many BTI researchers will present their latest research at the 13th annual SolGenomics Conference, Sept. 12-16 in Davis, California. Read more » -

The Watermelon’s Past, Present, and Future
Watermelons have changed from a small, bitter fruit that grows wild in Africa to the most popular fruit in the world. What's next for watermelons? Read more » -

BTI Scientists Envision the “Future of Food”
What will your dinner plate look like in 2050? With discoveries from the Boyce Thompson Institute, future crops may have more nutrients and greater resistance to insects, drought and disease. Read more » -

$6.5 Million Grant May Lead to Disease-Resistant Cucurbits
A consortium of 20 researchers is using advanced genomic techniques to accelerate the development of disease-resistant varieties of cucurbit crops. BTI Associate Professor Zhangjun Fei will lead the bioinformatics and genomics part of the initiative. Read more » -

From Flower to Fruit: Study Reveals Details of Tomato Formation
BTI Researchers pinpointed which genes are important at different stages of tomato fruit development by monitoring how gene expression changed in the first four days after a flower becomes pollinated. Read more » -

Student Symposium Caps Off 2015 Summer Intern Program
“My experience was really valuable...It confirmed the fact that I want to do science...science doesn’t work a lot of the time...it’s having the motivation and determination to tackle problems that you’re always going to come across.” Juan G Read more » -

BTI Welcomes New Crop of Summer Interns
BTI welcomes 20 college-level interns for 10 weeks of research in Plant Genome Research Program, the Bioinformatics Program or the Bioenergy Education Program. Read more » -

Extra DNA Creates Cucumber with All-Female Flowers
Researchers discover the genetics behind high-yield cucumbers that bear all-female flowers by screening 115 genome sequences to find large chromosomal variations. Read more » -
Scientists Work Together to Develop Global Distribution Map of Tomato Virus
Boyce Thompson Institute Scientist Zhangjun Fei has teamed up with scientists from across the country to generate a comprehensive global virus distribution map for tomatoes. Read more »
Internship Program | Projects & Faculty | Apply for an Internship
































Subscribe to BTI's LabNotes Newsletter!
Contact:
Boyce Thompson Institute
533 Tower Rd.
Ithaca, NY 14853
607.254.1234
contact@btiscience.org


Copyright © 2023 | Boyce Thompson Institute | All rights reserved | Privacy Policy | Cookie Policy