

Graduate Fields: Plant Biology
Plants respond to changes in their environment in a myriad of ways – some deeply conserved, and others more specific adaptations to a particularly harsh local environment. These responses to environmental stress can be complex, with copious interconnected regulatory mechanisms that contribute (hopefully) to a successful outcome. Work into understanding these regulatory mechanisms has largely focused on their protein components, and for good reason; functional and evolutionary analysis of proteins and the genes that encode them is relatively straightforward and these analyses have greatly expanded our understanding of how plants perceive and cope with stress.
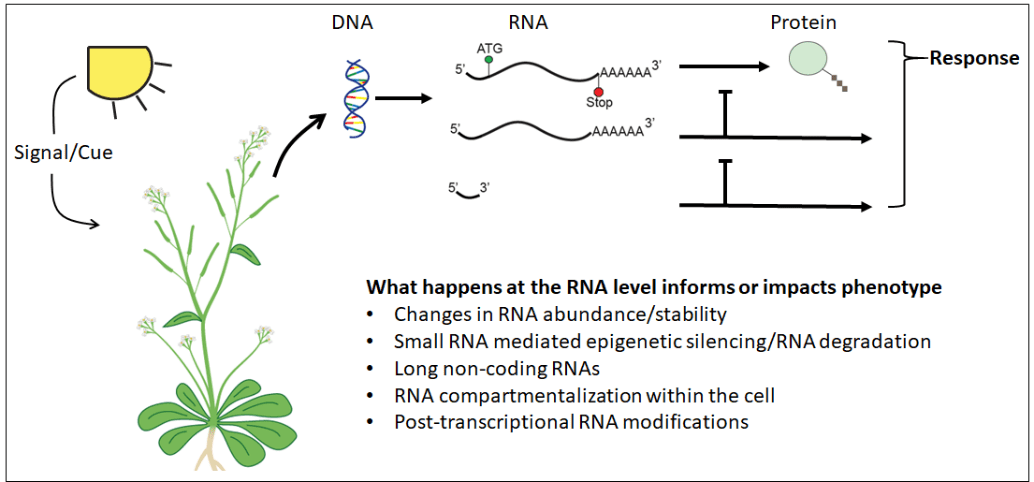 However, perception and regulation of stress occurs at the RNA level as well. This RNA-mediated regulation can occur through direct action of an RNA (micro and long non-coding RNA) on a target RNA/protein/DNA locus, or through chemical modification of RNAs (both mRNAs and ncRNAs) that change the target RNA’s structure, localization, or stability. These RNA-based regulatory mechanisms have the potential for both high specificity and sensitivity and therefore would be promising breeding targets in developing more stress-resilient crops. While promising, the who, where, when, and how of these RNA regulatory mechanisms are still poorly understood, particularly outside of Arabidopsis thaliana.
However, perception and regulation of stress occurs at the RNA level as well. This RNA-mediated regulation can occur through direct action of an RNA (micro and long non-coding RNA) on a target RNA/protein/DNA locus, or through chemical modification of RNAs (both mRNAs and ncRNAs) that change the target RNA’s structure, localization, or stability. These RNA-based regulatory mechanisms have the potential for both high specificity and sensitivity and therefore would be promising breeding targets in developing more stress-resilient crops. While promising, the who, where, when, and how of these RNA regulatory mechanisms are still poorly understood, particularly outside of Arabidopsis thaliana.
The Nelson lab uses comparative genomic, bioinformatic, and molecular approaches to better understand how plants regulate their stress responses at the RNA level and the degree to which these responses are conserved.
Current projects include:
The systematic identification and annotation of long non-coding RNAs in plants.
 The technology necessary to monitor gene expression in a single cell, within a tissue, or across an entire organism has developed tremendously over the past decade. As a direct result, there are now tens of thousands of publicly available data sets providing snapshots of how plants modulate the transcription of their genetic material to produce a phenotype. In order to appreciate the transcriptional complexity leading to phenotype, it is first necessary to understand the full composition of the transcriptome itself. Aside from protein-coding RNAs and small RNAs, a third class of transcript has recently been uncovered: long non-coding RNAs (lncRNAs). LncRNAs are emerging as key regulatory molecules impacting how plants respond to changes in their environment such as temperature and water abundance. Despite the their many important roles, lncRNAs remain poorly annotated in plants. LncRNAs are difficult to predict from genomic sequence alone and often require extensive transcriptional information, and the capacity to process that data, to confidently annotate. To overcome difficulties in lncRNA annotation and functional classification, this NSF-funded project is repurposing all (> 100 Tb) publicly available transcriptomic data for the top fifteen most studied model and agriculturally significant plant species (NSF-IOS PGRP 1758532). LncRNAs are being identified, cross-species conservation determined, and putative functional pathways inferred in each of the fifteen focal species.
The technology necessary to monitor gene expression in a single cell, within a tissue, or across an entire organism has developed tremendously over the past decade. As a direct result, there are now tens of thousands of publicly available data sets providing snapshots of how plants modulate the transcription of their genetic material to produce a phenotype. In order to appreciate the transcriptional complexity leading to phenotype, it is first necessary to understand the full composition of the transcriptome itself. Aside from protein-coding RNAs and small RNAs, a third class of transcript has recently been uncovered: long non-coding RNAs (lncRNAs). LncRNAs are emerging as key regulatory molecules impacting how plants respond to changes in their environment such as temperature and water abundance. Despite the their many important roles, lncRNAs remain poorly annotated in plants. LncRNAs are difficult to predict from genomic sequence alone and often require extensive transcriptional information, and the capacity to process that data, to confidently annotate. To overcome difficulties in lncRNA annotation and functional classification, this NSF-funded project is repurposing all (> 100 Tb) publicly available transcriptomic data for the top fifteen most studied model and agriculturally significant plant species (NSF-IOS PGRP 1758532). LncRNAs are being identified, cross-species conservation determined, and putative functional pathways inferred in each of the fifteen focal species.
Identification and characterization of stress-responsive and conserved base modifications
(aka the epitranscriptome) in plants.
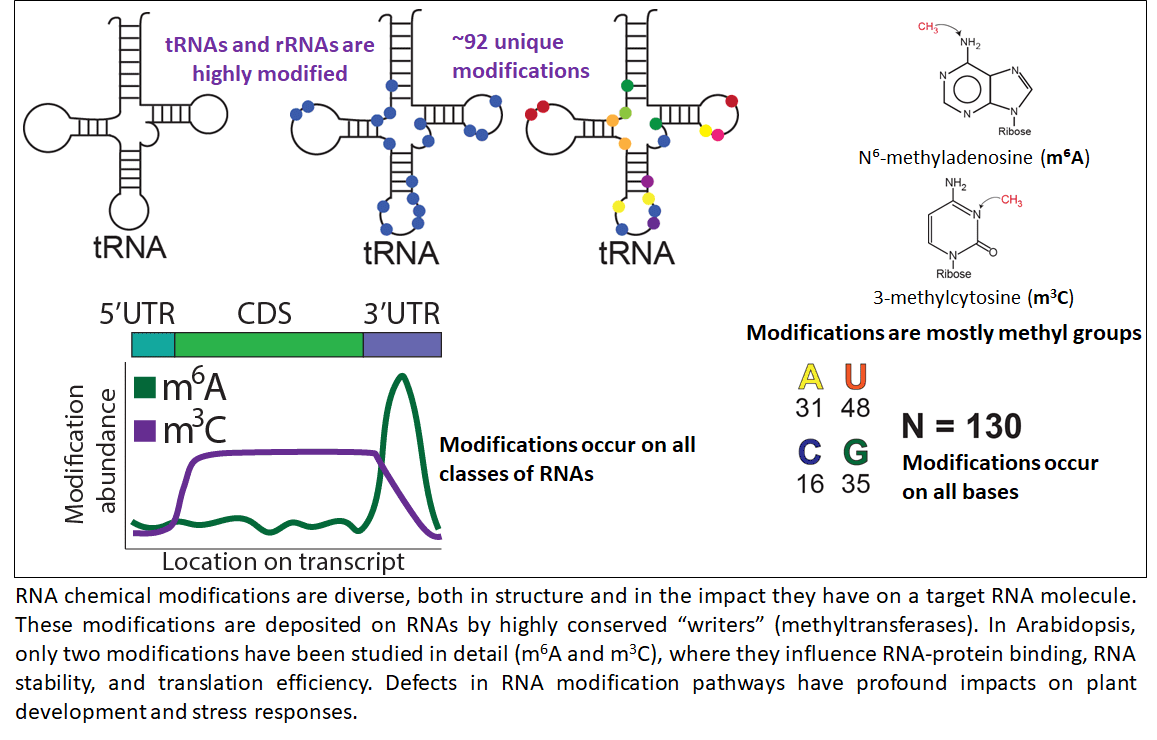
Across prokaryotes and eukaryotes, RNA chemical modifications are diverse, occur on all classes of RNA molecules, and are physiologically relevant. In plants, just two of the > 150 known RNA modifications have been studied in depth, and primarily in Arabidopsis, where they have been shown to have an impact at both the organismal and cellular levels. Little else is known about the role of the epitranscriptome in plants. This gap in knowledge is in large part due to the cost and technical difficulties of the biochemical assays used to measure abundance of specific RNA modifications. In light of these difficulties, in silico methods have been developed that facilitate high-throughput identification and prediction of these chemical additions. This NSF-funded collaborative project aims to address challenges in identifying modifications and placing them into a biological context by: 1) developing an exhaustive, annotated plant epitranscriptomic resource of over 47 unique modifications using approximately 1 petabase of publicly available RNA-seq data, and 2) provide a biological and evolutionary context for each of these modifications and the RNAs they are found to modify (NSF-IOS PGRP 2023310). To process the wealth of publicly available RNA-seq data and present the resulting information in a manner that will drive hypothesis generation, this project is developing novel computational workflows and data visualization tools. In sum, this project aims to provide the research community with an innovative plant epitranscriptome resource that is supplemented with sufficient biological and evolutionary context to facilitate in vivo functional analyses. This project is a collaboration with Drs. Brian Gregory (University of Pennsylvania), Eric Lyons (University of Arizona), and Rebecca Murphy (Centenary College of Louisiana).
Comparative analysis of stress-induced RNA modifications in the mustard (Brassicaceae) lineage.
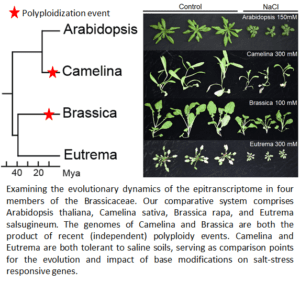 RNA sequencing has advanced dramatically in the past few years, with some platforms allowing researchers the ability to directly sequence RNA without the prior conversion to cDNA (what is sequenced by Illumina). Nanopore’s direct RNA sequencing captures not only an entire transcript but can also reveal if that transcript contains certain base modifications (i.e., m6A and m5C). We are applying this technology to address how these modifications influence transcriptional responses to heat and salt stress in four Brassicaceae: Arabidopsis thaliana, Camelina sativa, Brassica rapa, and Eutrema salsugineum. These four species represent an ideal comparative system. They reflect both diploid and recent polyploids (i.e., differing genomic influences on gene retention and function), as well as different levels of tolerance to these two stresses (e.g., Eutrema is highly salt tolerant whereas Arabidopsis is not). Finally, their evolutionary relationships to one another are well-described; Arabidopsis and Camelina reside in Lineage I of the family, whereas Brassica and Eutrema are in Lineage II, with the two lineages diverging from one another ~43 MYA. We are using this comparative system to better understand the degree to which m6A and m5C deposition during stress is conserved – especially between recently duplicated genes. In addition, we hope to understand how the evolution of RNA modifications influence stress tolerance or resilience in plants.
RNA sequencing has advanced dramatically in the past few years, with some platforms allowing researchers the ability to directly sequence RNA without the prior conversion to cDNA (what is sequenced by Illumina). Nanopore’s direct RNA sequencing captures not only an entire transcript but can also reveal if that transcript contains certain base modifications (i.e., m6A and m5C). We are applying this technology to address how these modifications influence transcriptional responses to heat and salt stress in four Brassicaceae: Arabidopsis thaliana, Camelina sativa, Brassica rapa, and Eutrema salsugineum. These four species represent an ideal comparative system. They reflect both diploid and recent polyploids (i.e., differing genomic influences on gene retention and function), as well as different levels of tolerance to these two stresses (e.g., Eutrema is highly salt tolerant whereas Arabidopsis is not). Finally, their evolutionary relationships to one another are well-described; Arabidopsis and Camelina reside in Lineage I of the family, whereas Brassica and Eutrema are in Lineage II, with the two lineages diverging from one another ~43 MYA. We are using this comparative system to better understand the degree to which m6A and m5C deposition during stress is conserved – especially between recently duplicated genes. In addition, we hope to understand how the evolution of RNA modifications influence stress tolerance or resilience in plants.
Understanding Mechanisms of Plant Resilience (alongside Magda Julkowska).
The Nelson and Julkowska labs make up the Mechanisms of Plant Resilience (MoPR) Cluster at BTI. The goal of the MoPR Cluster is to combine the strengths of the two labs (Julkowska = phenomics and stress physiology; Nelson = genomics and RNA biology) to develop a more complete understanding of the genetic and molecular factors associated with tolerance and acclimation to abiotic stress in domesticated plants and their wild relatives (primarily Solanaceae and Brassicaceae). Phenomics approaches will utilize both small scale but high temporal resolution Raspberry Pi imaging as well as large-scale high throughput phenotyping (HTP) platforms. Genomics approaches will span from single-cell RNA-seq to transcriptome-wide association studies (TWAS). Check back soon for more details!








Past Lab Members
 |
|---|




Subscribe to BTI's LabNotes Newsletter!
Contact:
Boyce Thompson Institute
533 Tower Rd.
Ithaca, NY 14853
607.254.1234
contact@btiscience.org


Copyright © 2023 | Boyce Thompson Institute | All rights reserved | Privacy Policy | Cookie Policy